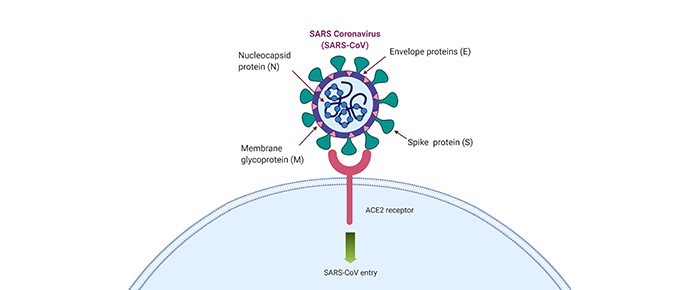
 Many problems are currently plaguing scientists studying COVID-19, as they try to solve why some patients suffer from more severe symptoms while others remain asymptomatic carriers. One of the pieces of that puzzle is the role of angiotensin-converting enzyme 2 (ACE2), the primary cell surface binding partner for SARS-CoV-2, which has yet to be fully elucidated.
Many problems are currently plaguing scientists studying COVID-19, as they try to solve why some patients suffer from more severe symptoms while others remain asymptomatic carriers. One of the pieces of that puzzle is the role of angiotensin-converting enzyme 2 (ACE2), the primary cell surface binding partner for SARS-CoV-2, which has yet to be fully elucidated.
What does ACE2 do?
ACE2 plays a major role in the renin-angiotensin system (RAS), which regulates blood volume, vascular resistance, cardiac output, and arterial blood pressure. Renin, which is secreted by the kidneys, is involved in the production of angiotensin II (AngII). When bound to angiotensin II type 1 receptor (AT1R), AngII induces vasoconstriction, cell proliferation, inflammation, and oxidative stress. However, AngII can also bind to angiotensin II type 2 receptor (AT2R), which has opposing effects.1
ACE2, a zinc metallopeptidase similar to ACE, is capable of hydrolyzing AngII, forming Ang(1-7). This neutralizes AngII signaling in two ways: it reduces the levels of AngII and elevates levels of Ang(1-7) eliciting a vasodilative, anti-fibrotic, and anti-inflammatory response.1,2 Following binding of ACE2 to AngII, ACE2 is either inactivated by AT1R-induced endocytosis, which prevents it from converting AngII into Ang1-7, or activated via AT2R, where it can convert AngII into Ang1-7.3
Outside of its role in the RAS, its activity has secondary effects in the lungs, as it regulates bradykinin 1 receptor signaling via inhibition of des-Arg9-bradykinin, thereby reducing vasodilation and vascular permeability. ACE is also expressed in enterocytes in the gut, where it can regulate amino acid homeostasis, antimicrobial peptide generation, local innate immunity, and microbial ecology.4,5
Though most have focused on ACE2’s role in the cardiovascular system, it’s found in a wide range of different tissues and cells types throughout the body:2
- Alveolar (type II) epithelial cells and pulmonary vasculature
- Goblet and ciliated epithelial cells in the upper airways
- Cardiofibroblasts, cardiomyocytes, endothelial cells, pericytes, and epicardial adipose cells in the heart
- Glomerular endothelial cells, podocytes, and proximal tubular endothelial cells in the kidneys
- Pigmented epithelial cells, rod and cone cells, and glial cells in the eyes
- Neurons and glial cells of the central nervous system6

ACE2 as a foe
It’s well known by now that SARS-CoV-2 uses ACE2 to enter cells, relying on TMPRSS2 for priming of the viral spike protein. The spike protein recognizes and binds to ACE2, allowing it to bind to the host’s plasma membrane and fuse with it.7
Some have suggested that high ACE2 levels are correlated with disease severity and infectivity due to its role as the virus’s primary entry point. This hypothesis has led several groups to measure the relative expression levels of ACE2 in different cohorts, including younger versus older individuals as the disease more frequently affects those aged 70 years or older and rarely affects children.
Bunyavanich et al recently attempted to determine why children aren’t as affected as older adults by measuring the levels of ACE2 in the nose. To do this, they took nasal epithelium samples collected from patients at the Mount Sinai Hospital Center in New York between 2015 and 2018. They then measured the mRNA levels of the ACE2 and grouped the data based on age, showing that levels were lower in children aged less than 10 years compared with adults.8
In a more thorough analysis, Smith et al looked at cigarette smoke, a risk factor for more severe COVID-19. They initially suggest that ACE2 levels don’t fluctuate much between different sexes or ages. However, when it came to smoking, the animals used in the study showed an 80% increase in ACE2 levels in the lung, and the human cohorts they studied showed that samples from smokers had around 30% to 55% more ACE2 expression compared with samples from non-smokers.9
In Smith’s paper, they indicate that the up-regulation of ACE2 is likely driven by the expansion of goblet cells, which can secrete mucous to help protect the respiratory tract, and that this response occurs in other respiratory diseases, such as chronic pulmonary obstructive disease (COPD) and during viral infection via inflammatory signaling.9
Finally, a recent paper by Hou et al found that the levels of both infectivity and ACE2 expression form a gradient across the various cells lining the respiratory tract, from the nose down to the lung. Because of this observation, they speculate that the nose represents the primary entry point of the virus, a finding that may strengthen Bunyavanich’s hypothesis that low ACE2 levels in the nose confer resistance to infection.10
ACE2 as a friend
Though the two studies listed above seem to correlate increased ACE2 expression with disease severity, they don’t provide a full picture. Many have pointed out that ACE2’s protective role in the lungs might serve as a double-edged sword in COVID-19: it helps the virus enter, but increasing its function might prevent the onset of severe disease.
Because ACE2 is responsible for regulating the levels of AngII, it’s been hypothesized that increased viral binding results in depletion of the receptor, leading to increased AngII levels, which can exacerbate symptoms, possibly by eliciting expression of cytokines like IL-6 and TNFα.11,12 Some small studies have already made a link, as patients with severe COVID-19 symptoms had high levels of plasma AngII, which correlated with high viral load and severe lung injury.11
It’s also unlikely that high ACE2 levels are the sole causative factor that results in disease severity, as certain diseases associated with worse outcomes, like hypertension, are also associated with increased AngII and low ACE2 expression and activity, particularly in adipocytes (fat cells). A similar trend might be present in endothelial cells, where upon entry of SARS-CoV-2, ACE2 loses its ability to prevent thrombosis.4 In these instances, low ACE2 levels likely wouldn’t protect from viral infection due to the high affinity of SARS-CoV-2 for ACE2.12
Because of the possible deleterious effects of increased AngII signaling, a few ongoing clinical trials are determining whether treatment with ACE inhibitors or AngII receptor blockers (ARBs) can affect the prognosis of patients with COVID-19 (NCT04318301, NCT04318418). There are also two studies assessing the effects of losartan, a nonpeptide AngII receptor antagonist, in patients with COVID-19 who are either hospitalized (NCT04312009) or ambulatory (NCT04311177).13
Soluble ACE2 in COVID-19?
Any talk of ACE2 has to mention that ACE2 can be cleaved in a process called “ectodomain shedding,” resulting in soluble ACE2 that can be detected in the blood.13 In this case, some believe that more soluble ACE2 would lead to worse prognosis in patients with COVID-19, resulting in less membrane-bound ACE2 and abolishing its ability to regulate AngII. This type of scenario, with high ACE2 plasma levels, occurs in both advanced heart failure and acute lung injury, and possibly COVID-19 as well.13
Why is ACE2 so important in COVID-19?
Understanding the role ACE2 plays in COVID-19 is not only critical for drug development but also for patients who rely on medications, like ARBs, for their normal health. During the pandemic, doctors must make snap judgments that might affect the outcome of the patient, and getting a full picture regarding the receptor dynamics and downstream signaling of ACE2 will be crucial to helping them make evidence-based choices.
LabTAG by GA International is a leading manufacturer of high-performance specialty labels and a supplier of identification solutions used in research and medical labs as well as healthcare institutions.

References:
- Sharma N, Anders HJ, Gaikwad AB. Fiend and friend in the renin angiotensin system: An insight on acute kidney injury. Biomed Pharmacother. 2019;110:764-774.
- Gheblawi M, Wang K, Viveiros A, et al. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ Res. 2020;126:1456–1474.
- Deshotels MR, Xia H, Sriramula S, Lazartigues E, Filipeanu CM. Angiotensin II mediates angiotensin converting enzyme type 2 internalization and degradation through an Angiotensin II type I receptor-dependent mechanism. Hypertension. 2014;64(6):1368-1375.
- Bourgonje A, Abdulle A, Timens W, et al. Angiotensin-converting enzyme-2 (ACE2), SARS-CoV-2 and Pathophysiology of Coronavirus Disease 2019 (COVID-19). J Pathol. 2020.
- Sodhi CP, Wohlford-Lenane C, Yamaguchi Y, et al. Attenuation of pulmonary ACE2 activity impairs inactivation of des-arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am J Physiol – Lung Cell Mol Physiol. 2018;314(1):L17-L31.
- Xia H, Lazartigues E. Angiotensin-converting enzyme 2 in the brain: Properties and future directions. J Neurochem. 2008;107(6):1482-1494.
- Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020:1-19.
- Bunyavanich S, Do A, Vincencio A. Nasal Gene Expression of Angiotensin-Converting Enzyme 2 in Children and Adults. JAMA. 2020.
- Smith J, Sausville E, Girish V, et al. Cigarette smoke exposure and inflammatory signaling increase the expression of the SARS-CoV-2 receptor ACE2 in the respiratory tract. Dev Cell. 2020;S1534-5807(20):30401-30409.
- Hou Y, Okuda K, Edwards C, et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell. 2020.
- Vaduganathan M, Vardeny O, Michel T, McMurray J, Pfeffer M, SD S. Renin–Angiotensin–Aldosterone System Inhibitors in Patients with Covid-1. N Engl J Med. 2020;382:1653-1659.
- Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020.
- Brojakowska A, Narula J, Shimony R, Bander J. Clinical Implications of SARS-Cov2 Interaction with Renin Angiotensin System. J Am Coll Cardiol. 2020;S0735-1097(20):35001-35004.



{kind=link}