

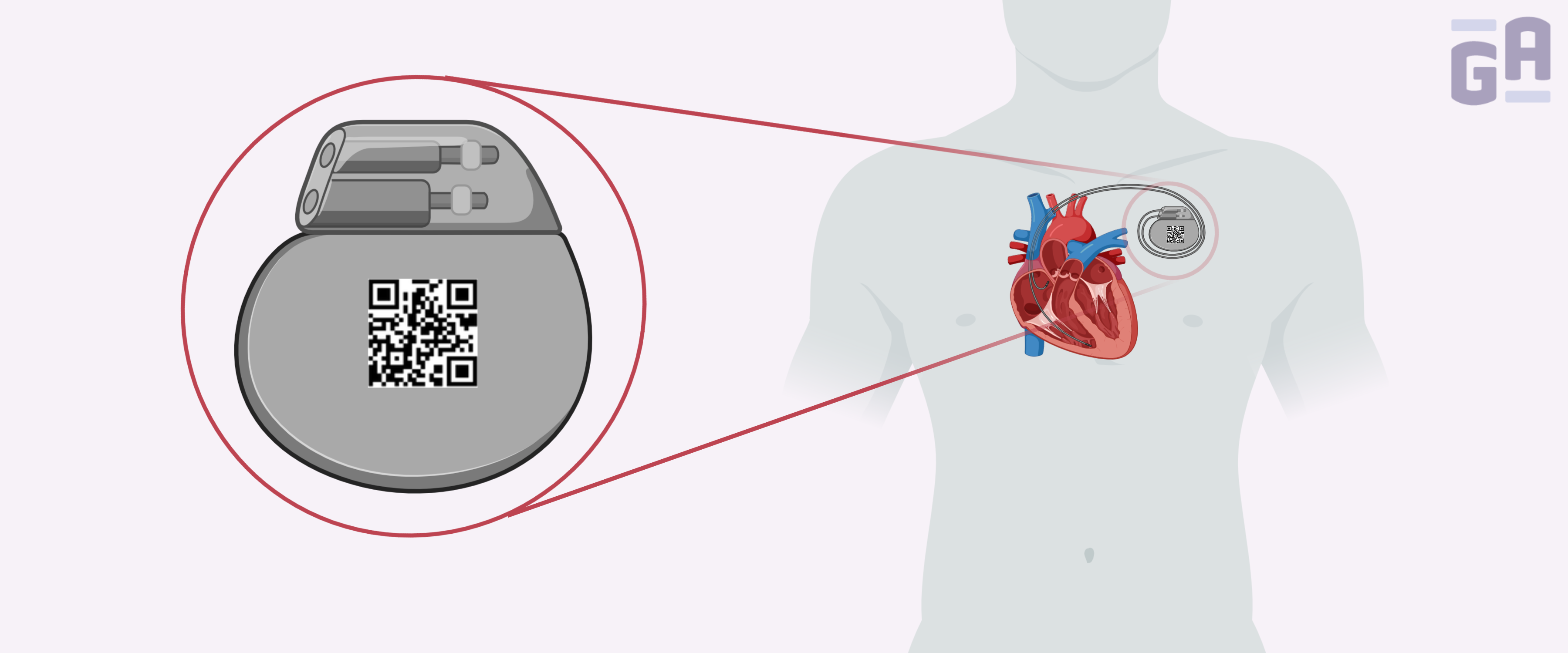

UDI regulations
UDIs are unique numeric or alphanumeric codes that consist of a device identifier (DI), which is a fixed identifier for the specific version or model of the device, and a production identifier (PI), which is a variable code that corresponds to either the lot, batch, serial number, expiration date, or date of production of the device. DI barcodes must be obtained from an FDA-accredited issuing agency, such as GS1, HIBCC, or ICCBBA.1
The FDA has set specific dates for when companies manufacturing medical devices must be fully compliant with the new regulations:2
- Currently, all Class II and III devices, which have a moderate to high risk for patients, require UDIs without exception.
- Class I devices that are not implantable, life-supporting, or life-sustaining devices will require UDIs by September 24, 2020.
- For Direct Mark (21 CFR 801.45) Class I devices, which are devices that require a permanent marking on the device itself due to its repeated use and reprocessing, the deadline is September 24, 2022.
Steps to complying with FDA’s UDI regulations3
- Obtain a DUNS number – To submit your UDIs to FDA’s Global Unique Device Identification Database (GUDID), your company will need a Data Universal Numbering System (DUNS) number.
- Identify the GMDN terms – To submit your UDIs to the GUDID, a Global Medical Device Nomenclature (GMDN) term code is needed for each device.
- Determine the number of UDIs required – Each model and version of the device will require its own UDI, including models that are differentiated only by color, size, style, material, or package size.
- Get your DIs – The DI portion of the UDIs is given by the FDA-accredited issuing agency.
- Choose a PI – UDIS for Class II and Class III medical devices must have a PI, which may include the lot/batch number, serial number, manufacturing date, or expiration date. For products that are human cell or tissue-based, they may require a distinct identification code.
- Pick a method for submitting your codes to GUDID – You can use FDA’s website interface or the Health Level 7 (HL7) option. While FDA’s interface only allows one submission at a time, HL7 allows many submissions simultaneously. However, pre-submission testing using a GUDID account must be completed prior to using HL7.
- Designate a regulatory contact – The regulatory contact will help assess all UDI requirements, set up the GUDID account, and act as a liaison between the company and the FDA.
- Create a GUDID account – This should only be performed once the company has confirmed how the data will be entered into the system. Once created, draft submissions can be a useful tool to familiarize yourself with the system and to help prepare the final submissions.
- Submit UDIs to GUDID – Gather all the information necessary, such as product descriptions, DIs, brand names, model/version numbers, GMDN codes, and FDA listing numbers, and submit.


How specialty labels can help
Using the right labels is crucial to comply with the FDA’s regulations on UDIs. Labels on medical devices that fall off or smudge can lead to a product that is no longer usable. There are several ways that specialty labels can help maintain compliance:
- Autoclave labels – Many devices, such as dental drills or any device that is implanted into the body, must be sterilized prior to use. The most common sterilization method is steam autoclaving at temperatures up to 120°C and pressures up to 30 psi. For these devices, it’s important to use autoclave labels. These labels will withstand the elevated temperature and pressure of autoclaving in addition to gamma irradiation (up to 50 KGy) and exposure to ethylene oxide gas, both of which may also be used to sterilize devices.
- Chemical-resistant labels – Often, devices need to be sterilized with disinfectants. For example, IV pumps need to be cleaned with sanitizing wipes regularly. Chemical-resistant labels can withstand long-term exposure to most disinfectants, including ethanol (up to 100%), isopropanol, and bleach, ensuring they remain affixed to your device and legible, no matter how many times they’re cleaned.
- Cryogenic labels – Some types of devices are based on human tissues and cells, such as cultured cells for regenerative therapy and collagen. These devices may require storage in liquid nitrogen (-196°C), making cryo labels the ideal identification solution, as they remain affixed during long-term cryogenic storage.
The printout is just as important as the label itself. No matter what environment your medical devices encounter, using a thermal-transfer printer confers optimal protection against smudging and fading.
Though the FDA has given companies ample time to adapt to the new regulations, it may still be a challenge to achieve full compliance over the long term. Applying the right labels and printing method is an easy first step towards fulfilling these requirements that can be implemented relatively quickly. It can also help overcome other healthcare-related obstacles, such as maintaining barcode readability, which is necessary for integrating medical devices into electronic health reporting systems.4 Overall, using the appropriate labels will help ensure your company is set up to meet all the FDA’s requirements on UDIs.
LabTAG by GA International is a leading manufacturer of high-performance specialty labels and a supplier of identification solutions used in research and medical labs as well as healthcare institutions.


References:
- US Food and Drug Administration (FDA). UDI Basics. Silver Spring, MD; 2018.
- US Food and Drug Administration (FDA). FDA Letter of Intent to Extend UDI Class I and Unclassified Devices. Silver Spring, MD; 2017.
- Corp R. Ten Steps to Comply With FDA UDI Requirements. https://www.registrarcorp.com/comply-fda-udi-top-ten/. Published 2015. Accessed August 5, 2019.
- Campion TR, Johnson SB, Paxton EW, Mushlin AI, Sedrakyan A. Implementing unique device identification in electronic health record systems: Organizational, workflow, and technological challenges. Med Care. 2014;52(1):26-31.



{kind=link}