

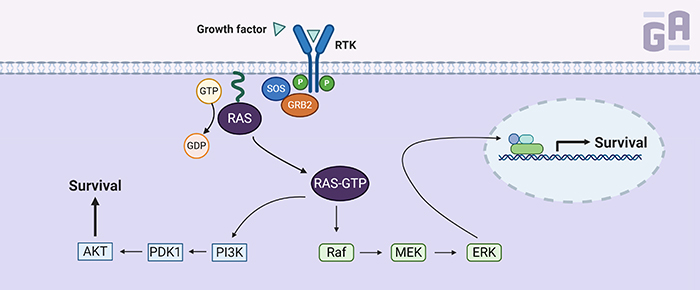

What is K-Ras, & why is it so hard to target?
K-Ras is one of three RAS genes found in humans—the others being NRAS and HRAS—that encode small guanosine triphosphatases (GTPases), which convert GTP to GDP (small signaling molecules). K-Ras normally resides in a GDP-bound state, rendering it inactive. It requires a guanosine nucleotide exchange factor (GEF) to remove the GDP and replace it with GTP, activating the protein and causing a signaling cascade involving multiple pathways that often result in gene expression related to growth, proliferation, and survival.
So, why is it so hard to target K-Ras directly? K-Ras has an extremely high affinity for GTP (in the picomolar range) to go along with millimolar concentrations of it surrounding the protein, so early attempts at designing drugs to target K-Ras activation often failed because GTP would outcompete the drug. Indirect methods of targeting the K-Ras protein were adapted instead, including drugs that prevented it from sticking to the plasma membrane. By targeting farnesyl transferase, an enzyme that adds a lipid modification to K-Ras, scientists were able to block its association with the membrane; however, it had little effect in humans when clinically tested in phase III trials. Two other molecules that disrupt the localization of K-Ras through inhibiting prenylation (the addition of hydrophobic modifications), deltarasin and deltazinone, have also been evaluated in experimental models of Ras-driven cancer. However, both have yet to be tested clinically.1
Another method that’s been used to block K-Ras signaling is by targeting its downstream effectors. Several clinical trials currently underway for inhibitors of molecules involved in the K-Ras signaling cascade, such as RAF, Mek, and Erk. However, these effectors are not so easy to pinpoint, as inhibition of one molecule can often lead to the activation of a parallel pathway that bypasses the cascade and keeps the tumor growing. This has led to several ongoing trials using a combination of inhibitors for at least 2 pathways, such as RAF and PI3K. Some have even tried using a combination of anti-Ras drugs with inhibitors of downstream signaling proteins.2


A new class of small molecules
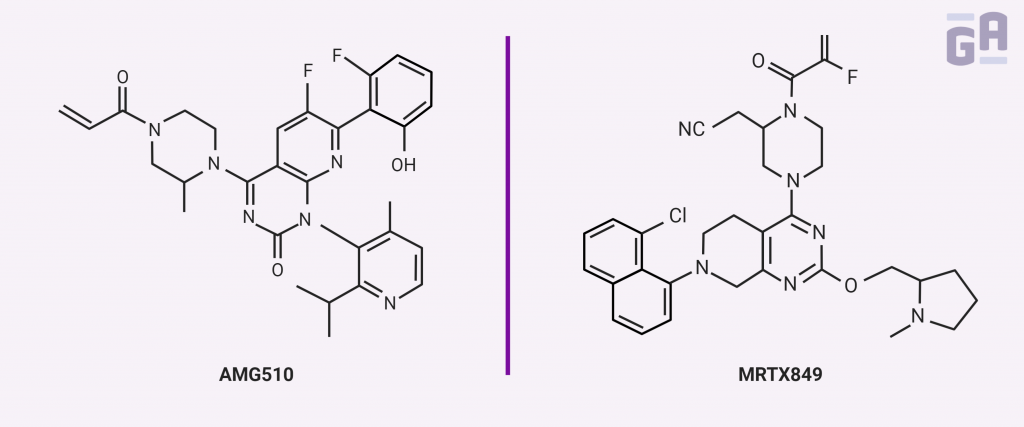
One of the greatest breakthroughs of 2019 in pharma was that K-Ras’s hyperactive GTPase activity could be directly targeted using small molecule inhibitors. Two groups recently reported molecules that could directly bind mutant forms of K-Ras with enough specificity and sensitivity to give scientists hope that these could be potential upgrades over current treatments for Ras-driven cancers.
One of the two groups includes Amgen, who treated both animals and human patients with a compound called AMG 510. This small molecule covalently binds to a cryptic surface groove in the GDP-bound form of the K-Ras G12C mutant, recognizing the mutated protein but not the wild-type form. In vitro, it prevented the exchange of GDP for GTP relatively efficiently and inhibited downstream signaling through ERK. In mice, AMG 510 was effective at reducing tumor volume only when an active immune system was present, which led to further experimentation using immunotherapy with antibodies directed against the immune-modulating protein, programmed cell death-1 (PD-1), that often allows tumors to escape immune surveillance. The combination of AMG 510 and PD-1 antibodies was better than either treatment individually, curing 90% of immune-competent mice and even making them resistant to the reintroduction of tumorigenic cells. Amgen also had success in patients with non–small cell lung cancer (NSCLC), with four patients who all showed positive responses to the drug.3
One other group had some clinical success directly targeting K-Ras by stabilizing the GDP-bound form. In a paper by Hallin et al. in Cancer Discovery, they describe a small molecule inhibitor, MRTX849, that selectively repressed mutant K-Ras activity in both pancreatic and lung cancer cell lines as well as in patient xenograft and cell line-derived mouse models, where it elicited tumor regression in 17 of the 26 models tested. The drug was further tested in at least two patients with lung cancer who no longer responded to conventional chemotherapies, with each showing a partial reduction in tumor growth. The authors also suggested that combining MRTX849 with both upstream and downstream signaling inhibitors could further boost its anticancer activity.4

Directly targeting KRAS with RNAi
RNA interference (RNAi) techniques can be used to silence gene expression. At least one group, working out of the MD Anderson Cancer Center, has used siRNA—an RNAi method that utilizes double-stranded RNA molecules—to effectively silence KRAS gene expression, thereby directly inhibiting its ability to signal. Unlike many siRNA therapeutics delivered via lipid nanoparticles, Dr. Raghu Kalluri and his group used exosomes, endogenous extracellular vesicles that consist of a lipid bilayer and measure around 40 to 150 nm in size, to deliver KRAS-specific siRNAs. They purified the exosomes, electroporated them to include the siRNAs, and injected them back into mice harboring pancreatic tumors resistant to traditional chemotherapeutic agents. Using this strategy, they were able to lower K-Ras expression levels, reduce the size of the tumors, and inhibit K-Ras–associated downstream signaling.5 This therapy is now entering phase I clinical trials for patients with pancreatic cancer and a KRAS G12D mutation (NCT03608631).6
Another form of RNAi used to target KRAS is microRNAs (miRNAs), which are short non-coding RNAs 22 to 24 base pairs long that target the 3ʹ untranslated region of mRNAs, effectively inhibiting translation. One paper, published by Mario Acunzo et al. in the Proceedings of the National Academy of Sciences of the United States of America (PNAS), designed a miRNA specific for point-mutated KRAS transcripts and tested their construct in vitro with cell line models of lung cancer. Their KRAS miRNA not only inhibited mutant K-Ras downstream activity, but it also made the cells sensitive to other chemotherapeutics, such as the epidermal growth factor receptor inhibitor gefinitib, without inducing any observable off-target effects.7
K-Ras biologics
Antibody targeting of K-Ras represents yet another method of reducing its activity. One paper, from Kim et al. in 2017, described an antibody that bound only to the active form of Ras, and that was combined with the backbone of an antibody (TMab4) capable of penetrating cells. Some have even used antibody mimetics in colon cancer models that target GDP-bound Ras to prevent nucleotide exchange and effectively inhibit signaling through both Erk and Akt downstream pathways.8


References:
- Porru M, Pompili L, Caruso C, Biroccio A, Leonetti C. Targeting kras in metastatic colorectal cancer: Current strategies and emerging opportunities. J Exp Clin Cancer Res. 2018;37(1):1-10.
- Misale S, Fatherree JP, Cortez E, et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin Cancer Res. 2019;25(2):796-807.
- Herbst RS SJ. Small molecule combats cancer-causing KRAS protein at last. Nature. 2019.
- Hallin J, Engstrom LD, Hargis L, et al. The KRASG12C Inhibitor, MRTX849, Provides Insight Toward Therapeutic Susceptibility of KRAS Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2019.
- Kamerkar S, Lebleu VS, Sugimoto H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546(7659):498-503.
- Setten RL, Rossi JJ, Han S ping. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18(6):421-446.
- Acunzo M, Romano G, Nigita G, et al. Selective targeting of point-mutated KRAS through artificial microRNAs. Proc Natl Acad Sci U S A. 2017;114(21):E4203-E4212.
- Spencer-Smith R, O’Bryan JP. Direct inhibition of RAS: Quest for the Holy Grail? Semin Cancer Biol. 2019;54:138-148.




{kind=link}